Publication Highlight: The New Wave of Closing Genomes!
It seems like only yesterday that marker gene analysis was the only feasible option available for assessing the microbial content of samples. Whole metagenome shotgun sequencing has since taken the field by force. The more data that we acquire, the more we want to extract from it. Closing genomes seems to be the next item on the agenda! A recent publication from Eli Moss, Dylan Maghini, and Ami Bhatt1 describes their great work to get the most out of their data, and their approach to closing genomes from complex microbiome samples.
Assembling complete genomes from metagenomic sequence data comes with quite a few challenges. If using short reads only, assembling large contiguous sequences can be hindered by repetitive elements. Horizontal gene transfer events can be missed and lead to incorrect assemblies. Long read technology can help overcome some of these problems, but comes with its own caveats. Long read technologies tend to have higher error rates, and high molecular weight DNA is required to get reliable long reads. When dealing with multi-organism samples such as stool microbiomes, extracting DNA becomes key. If you’re too gentle, you don’t break open the hardy microbes, leaving you with a bias in abundance representation. If you’re too vigorous, you sheer the DNA, making it more difficult to obtain long reads.
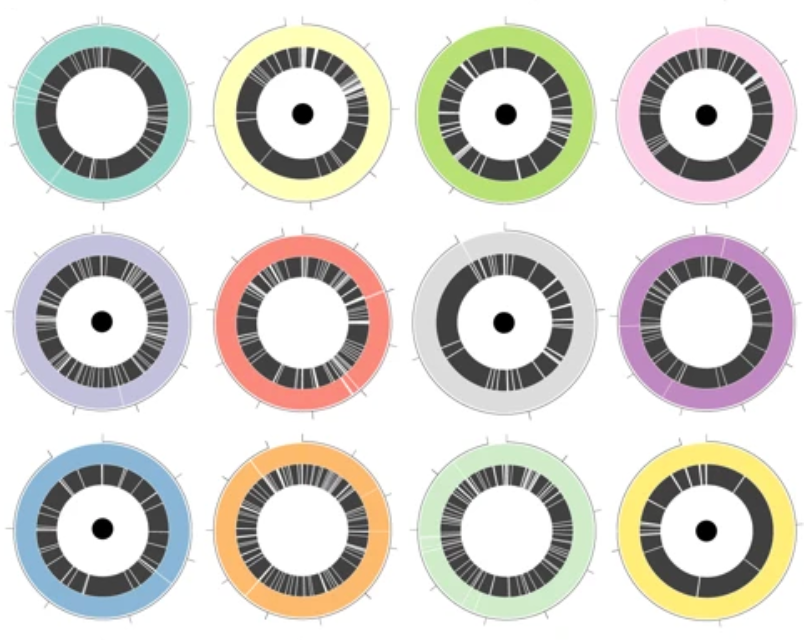
Fig. 1: Circos plots from Moss et al. (2020), showing contiguity of Nanopore assemblies (outer rings) compared to short-read assemblies (inner rings), with black dots indicating complete genomes.
The Bhatt Lab attempted to tackle these issues with a multi-pronged approach. They optimized nucleic acid extraction techniques for high molecular weight DNA, allowing them to perform long read sequencing with Oxford Nanopore Technology’s MinION. They coupled this with a bioinformatics workflow they developed, named Lathe. Lathe uses Flye2 or Canu3, two state of the art long-read assemblers, to assemble genomes directly from metagenomic read sets. Lathe was able to obtain higher N50 scores than other long-read assembly tools on stool microbiome samples. Using a synthetic cocktail of 12 known species, their workflow was able to assemble seven of those genomes into single and complete contigs, and a further three genomes were assembled into four contigs or fewer. When applied to human stool samples, one of the genomes they succeeded in closing was Prevotella copri; a microbe that has been very challenging to close for many in the field. It was only recently closed by Stewart et al. (2019)4. Another recent publication by Bertrand et al. (2019)5 presented a hybrid-assembly tool, OPERA-MS, which also contributes to solving some of the issues discussed above.
We at One Codex are also excited about closing genomes! In a recent blog, we presented the ATCC Genome Portal, highlighting our efforts with the ATCC to close and fully circularize bacterial genomes from ATCC’s collection. We use a hybrid assembly approach, where long reads provide scaffolding, and short reads provide accuracy. We then perform rigorous quality control analyses on the assemblies, to ensure that we contribute only the highest-quality genomes to the field. And we’re eager to continue to grow our databases with complete genomes!
If you have any questions about the ATCC Genome Portal, you can reach out to us at hello@onecodex.com or use the chat at the bottom right of your screen.
References:
1. Moss, E.L., Maghini, D.G. & Bhatt, A.S. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat Biotechnol (2020). https://doi.org/10.1038/s41587-020-0422-6
2. Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol (2019). https://doi.org/10.1038/s41587-019-0072-8
3. Koren, S., Walenz, B.P., Berlin, K., Miller, J.R., Bergman, N.H. & Phillippy, A.M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res (2017). https://doi.org/10.1101/gr.215087.116
4. Stewart, R.D., Auffret, M.D., Warr, A., Walker, A.W., Roehe, R. & Watson, M. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat Biotechnol (2019). https://doi.org/10.1038/s41587-019-0202-3
5. Bertrand, D., Shaw, J., Kalathiyappan, M., Ng, A.H.Q., Kumar, M.S., Li, C., Dvornicic, M., Soldo, J.P., Koh, J.Y., Tong, C., Ng, O.T., Barkham, T., Young, B., Marimuthu, K., Chng, K.R., Sikic, M. & Nagarajan, N. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat Biotechnol (2019). https://doi.org/10.1038/s41587-019-0191-2