Running new analyses & whole-genome alignments
Today we’d like to tell you about a new feature on One Codex that allows you to run new analyses against your samples, including AMR gene panels and whole-genome alignments.
Running New Analyses
When samples are uploaded to One Codex, we automatically classify them using the One Codex Database of ~40K complete microbial genomes. However, metagenomic classification is just one of a range of microbial analysis tools provided by the One Codex platform. While in the past we’ve configured additional analyses to run automatically where appropriate (e.g., MLST for common bacterial isolates) or at the request of users, our new Run Analysis page allows you to run an in silico panel or perform whole-genome alignments against any of your samples.
To start a new analysis go to the Run Analysis page (shown below), where you can select one or more samples by using the search bar on the left. Once you have a set of samples selected, just select an analysis to perform on the right and click “Run”.
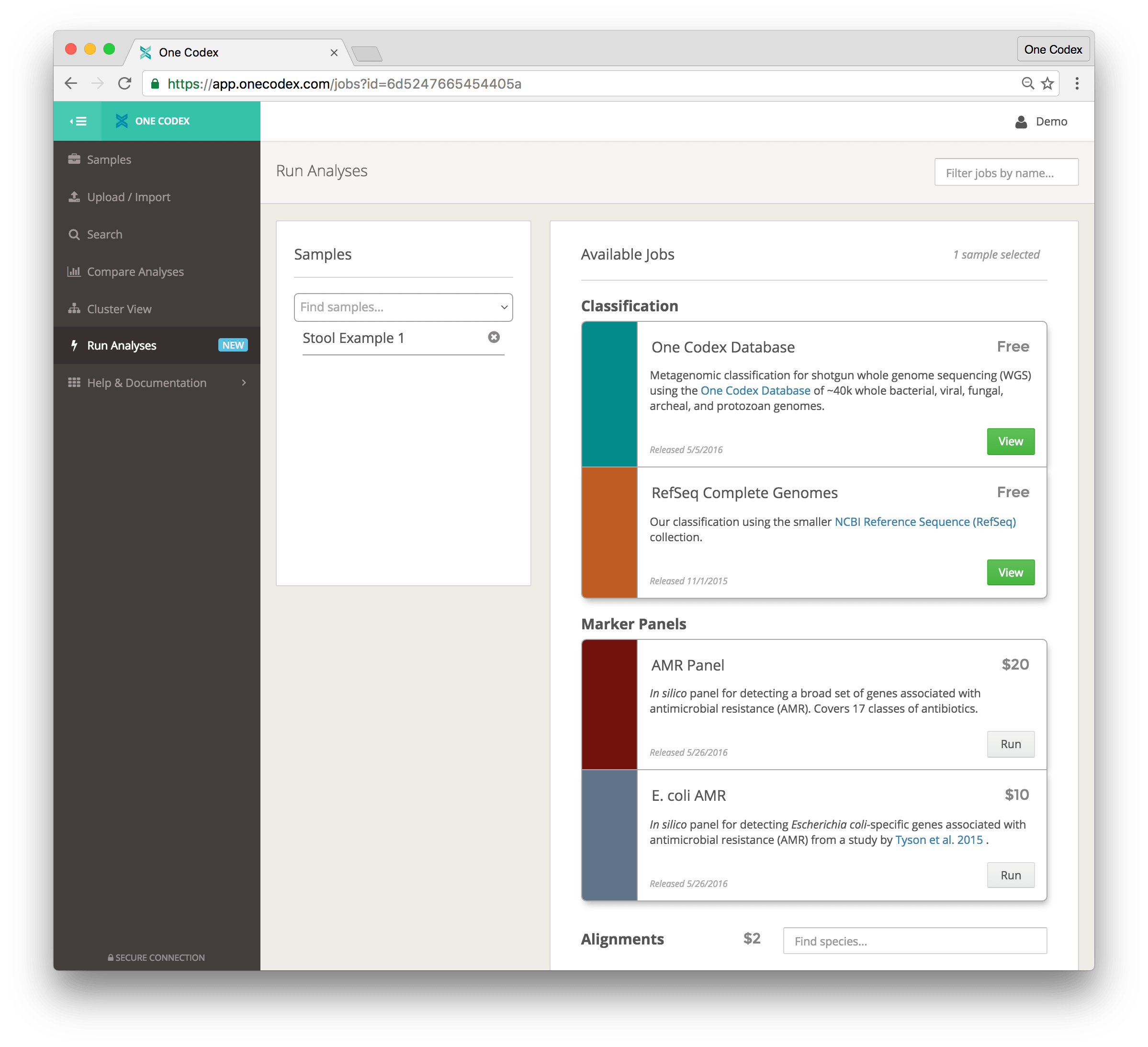
Classification analyses detect broad sets of microbes across our reference databases, while Marker Panels characterize specific genes or SNPs with known functions (such as Anti-Microbial Resistance genes in E. coli). In addition, you can perform Whole-Genome Alignment against any species or strain of interest from the One Codex Database.
Whole-Genome Alignments
While metagenomic classification can quickly and accurately pick out the taxa present in a sample, whole-genome alignment is an orthogonal tool for evaluating the presence, identity, coverage, and sequencing depth of an individual strain or species in a sample. This is particularly useful when it’s important to quantify relative coverage of an organism or similarity to a specific reference genome.
Below is an example of a stool microbiome sample that has been aligned against the three most abundant species from the metagenomic classification results:
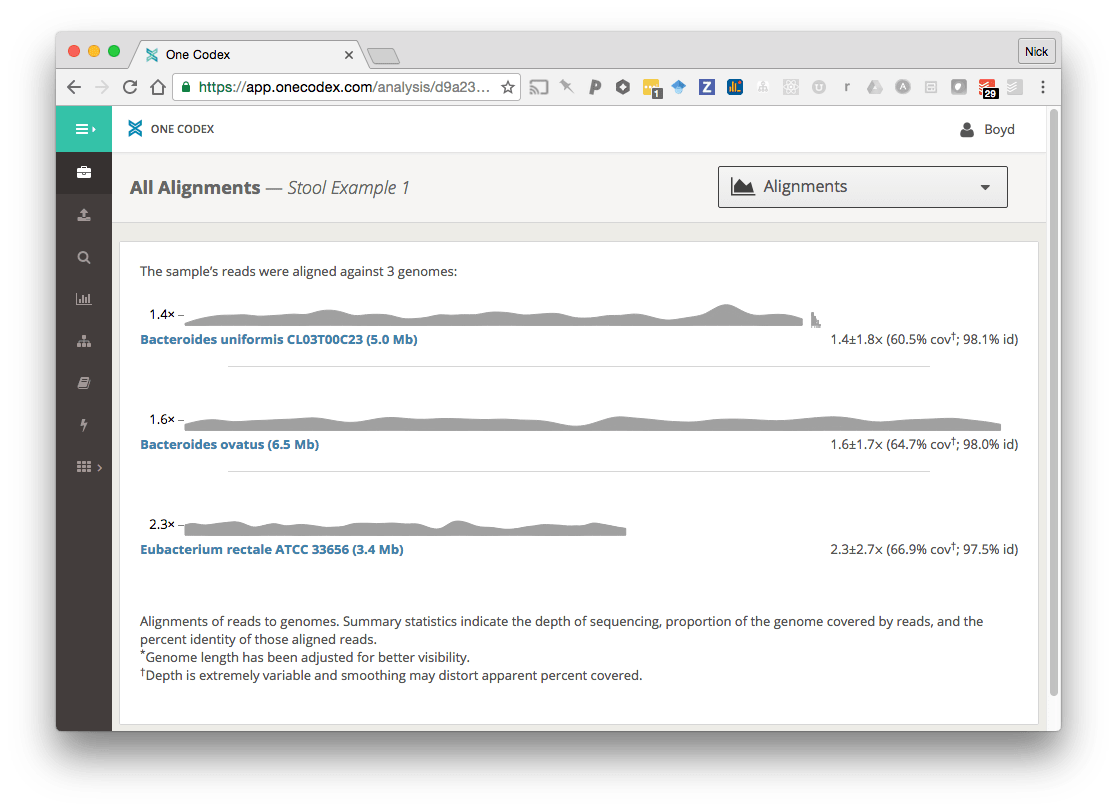
In addition to the coverage plot showing where the reads align across the genome, we calculate a set of summary statistics:
- Sequencing depth: The average number of reads piled up at every position in the genome
- Genome coverage: The percentage of the genome that is covered by the reads or contigs
- Nucleotide identity: The similarity of the sample to the reference genome (including SNPs and insertion/deletions)
The ability to run new analyses, including whole-genome alignments, expands the range of tools available to users on the One Codex platform. We expect to continually expand this toolbox with more organism-specific and functional analyses over time.
Please drop us a note if there’s an analyses you’d love to see supported or we can help in any other way.
– The One Codex Team